
![[ TSBT's Pirate ]](./images/ranks/British Army Ranks/Small Ranks/17_General.gif "[ TSBT's Pirate ]")
By: The FightAIDS@Home research team
15 Jun 2017
Summary
FightAIDS@Home researchers restarted the first phase of the project at the end of 2016, and in just a few months, they have completed approximately 46 percent of their projected work on World Community Grid. Read about their progress on finding compounds that could stop HIV from replicating.
Background
FightAIDS@Home is searching for possible compounds to target the protein shell of HIV (called a capsid), which protects the virus. Currently, there are no approved drugs that target this protein shell.
The virtual docking techniques used in Phase 1 are an approximation of the potential effectiveness of promising compounds. Phase 2 of FightAIDS@Home uses a different simulation method to double-check and further refine the virtual screening results that are generated in Phase 1.
The research team is examining a library of approximately 1.6 million commercially available compounds to find promising treatment prospects. The team estimates that they will need to carry out roughly 621 million docking computations on World Community Grid to thoroughly test each potential compound. With the help of many volunteers who are supporting this project, they’ve already completed 46 percent of their goal.
You can keep up with the research team’s progress on their website, which includes frequent updates on their experiments and progress.
Please read below for a detailed look at the technical aspects of their recent work.
Insilico search for novel drugs targeting the HIV-1 mature capsid protein
The importance of the capsid protein
The capsid protein (CA) plays crucial roles in the HIV replication cycle1. After viral and host cell membrane fusion, the capsid core is released into the cytoplasm. This core, which corresponds to the assembly of ~1200 capsid proteins, contains and protects viral RNA and proteins from degradation. Reverse transcription occurs in the core in a process which is tightly connected to the capsid core disassembly. This leads to the import of the cDNA viral genome into the host cell’s nucleus, where it is integrated into the host DNA to finalize the infection.
To date, no drugs targeting CA are approved for clinical use. With the goal of identifying novel active molecules which destabilize the capsid core, we set up a high throughput virtual screening (VS) campaign in collaboration with World Community Grid as part of the FightAIDS@Home (FA@H) project.
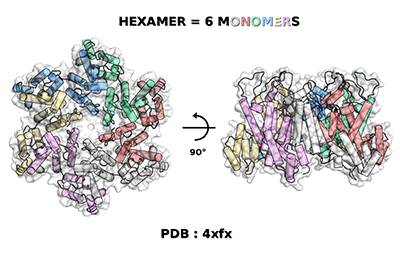
Figure 1: PDB 4xfx, the hexamer structure of the native HIV-1 mature capsid protein. (Credit: Pierrick Craveur)
Targeted structures
The main target of the docking calculations was the recently solved structure of the CA hexameric assembly2. Four pockets of interest were selected at the surface of the hexamer in order to perform focused dockings, mainly at the CA-CA dimer interfaces. Structural variability surrounding these pockets was analyzed by comparing this X-ray structure from the PDB (4xfx, see Figure 1), and the two full capsid core models assembled by Schulten's lab3 (3j3q and 3j3y, see Figure 2). Based on that, 36 different conformations were selected as targets for the VS, including the X-ray structure and structures from the models. Each target was set as full rigid and also with a specific combination of residue side chains defined as flexible.
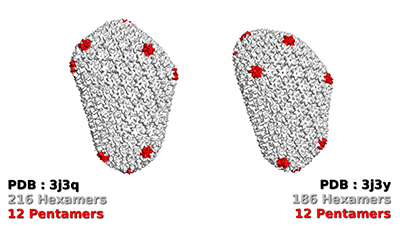
Figure 2: The 2 models of the capsid core assembly. (Credit: Pierrick Craveur)
An extended library of ~1.6 million commercially available compounds was used for the screening. Replicate computations were performed for each docking experiment in order to assess the consistency of the results. In total ~621 million docking computations will be performed on World Community Grid. For the time being, ~46% of the computation is completed, with an ending date estimated at the end of 2017 if the computation does not increase in speed. However, in one month we will be able to propose to our collaborators from the HIVE Center a selection of compounds (focusing one of the four pockets) for experimental binding and infectivity assays.
Other information
Dedicated web pages (see fightaidsathome.scripps.edu/Capsid/index) were developed to inform the public and the World Community Grid volunteers as the project advances. The pages contain an overview of the project, details on targets and the selection process, a description of the compound library, an hourly updated status of the computations, and a “people” section where volunteers can appear in the page to be fully part of the project.
An automatic pipeline has been developed in order to constantly post-process the docking results received from World Community Grid. These post computations involve the High Performance Computing (HPC) cluster from The Scripps Research Institute, and are mainly related to the identification of the interactions between drug candidates and the CA protein. The pipeline ends in filling a MySQL database, which will be made public as soon as it will be stable. In details, 3.3TB of compressed data are estimated to be received from World Community Grid, and 1TB to be generated after post-processing.
Our team from The Scripps Research Institute of San Diego, which includes Dr. Pierrick Craveur, Dr. Stefano Forli, and Prof. Arthur Olson, really appreciates the essential support this project receives from World Community Grid volunteers around the globe.
References
1. Campbell, E. M. & Hope, T. J. HIV-1 capsid: the multifaceted key player in HIV-1 infection. Nat Rev Microbiol 13, 471-483, doi:10.1038/nrmicro3503 (2015).
2. PDB 4xfx : Gres AT, Kirby KA, KewalRamani VN, Tanner JJ, Pornillos O, Sarafianos SG. X-Ray Structures of Native HIV-1 Capsid Protein Reveal Conformational Variability. Science (New York, NY). 2015;349(6243):99-103.
3. PDB 3j3q & 3j3y : Zhao G, Perilla JR, Yufenyuy EL, et al. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature. 2013;497(7451):643-646.
